bash
/opt/cellranger/8.0.1/lib/bin/bamtofastq \
--nthreads=8 \
--cr11 \
/datapool/datasets/pbmc3k/downloaded/pbmc3k_possorted_genome_bam.bam \
/datapool/datasets/pbmc3k/fastqsbash
/opt/cellranger/8.0.1/lib/bin/bamtofastq \
--nthreads=8 \
--cr11 \
/datapool/datasets/pbmc3k/downloaded/pbmc3k_possorted_genome_bam.bam \
/datapool/datasets/pbmc3k/fastqsbamtofastq v1.4.1
WARNING: no @RG (read group) headers found in BAM file. Splitting data by the GEM well marked in the corrected barcode tag.
Reads without a corrected barcode will not appear in output FASTQs
Unrecognized 10x BAM file.
For BAM files produced by older pipelines, use one of the following flags:
–gemcode BAM files created with GemCode data using Longranger 1.0 - 1.3
–lr20 BAM files created with Longranger 2.0 using Chromium Genome data
–cr11 BAM files created with Cell Ranger 1.0-1.1 using Single Cell 3’ v1 data
bash
/opt/cellranger/8.0.1/bin/cellranger count \
--id=pbmc3k \
--transcriptome=/datapool/reference_genomes/gencode/human/47/index/cellranger/GRCh38 \
--fastqs=/datapool/datasets/pbmc3k/fastqs/gemgroup001 \
--output-dir=/datapool/datasets/pbmc3k/alignments \
--localcores=8 \
--localmem=60 \
--create-bam true \
--nosecondaryR
library(Shennong)
library(Seurat)Loading required package: SeuratObjectLoading required package: sp'SeuratObject' was built with package 'Matrix' 1.7.1 but the current
version is 1.7.2; it is recomended that you reinstall 'SeuratObject' as
the ABI for 'Matrix' may have changed
Attaching package: 'SeuratObject'The following objects are masked from 'package:base':
intersect, tlibrary(tidyverse)── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
✔ dplyr 1.1.4 ✔ readr 2.1.5
✔ forcats 1.0.0 ✔ stringr 1.5.1
✔ ggplot2 3.5.1 ✔ tibble 3.2.1
✔ lubridate 1.9.4 ✔ tidyr 1.3.1
✔ purrr 1.0.4 ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errorsoutdir <- "/datapool/datasets/pbmc3k/analysis"
dir.create(path = outdir, recursive = TRUE)Warning in dir.create(path = outdir, recursive = TRUE): cannot create dir
'/datapool', reason 'Permission denied'counts <- sn_read("https://zenodo.org/records/14868137/files/filtered_feature_bc_matrix.h5?download=1")
seurat_obj <- sn_initialize_seurat_object(
x = counts,
project = "pbmc3k", species = "human"
)Maps last updated on: Sat Nov 16 10:35:32 2024Warning in HGNChelper::checkGeneSymbols(x = rownames(counts), species =
species): Human gene symbols should be all upper-case except for the 'orf' in
open reading frames. The case of some letters was corrected.Warning in HGNChelper::checkGeneSymbols(x = rownames(counts), species =
species): x contains non-approved gene symbolsWarning: Different cells and/or features from existing assay RNAMaps last updated on: Sat Nov 16 10:35:32 2024
Maps last updated on: Sat Nov 16 10:35:32 2024Warning in HGNChelper::checkGeneSymbols(genes, species = species): x contains
non-approved gene symbolsseurat_obj <- sn_filter_cells(x = seurat_obj,
features = c("nFeature_RNA", "nCount_RNA",
"percent.mt"))Warning: Overwriting miscellanous data for qc
Warning: Overwriting miscellanous data for qc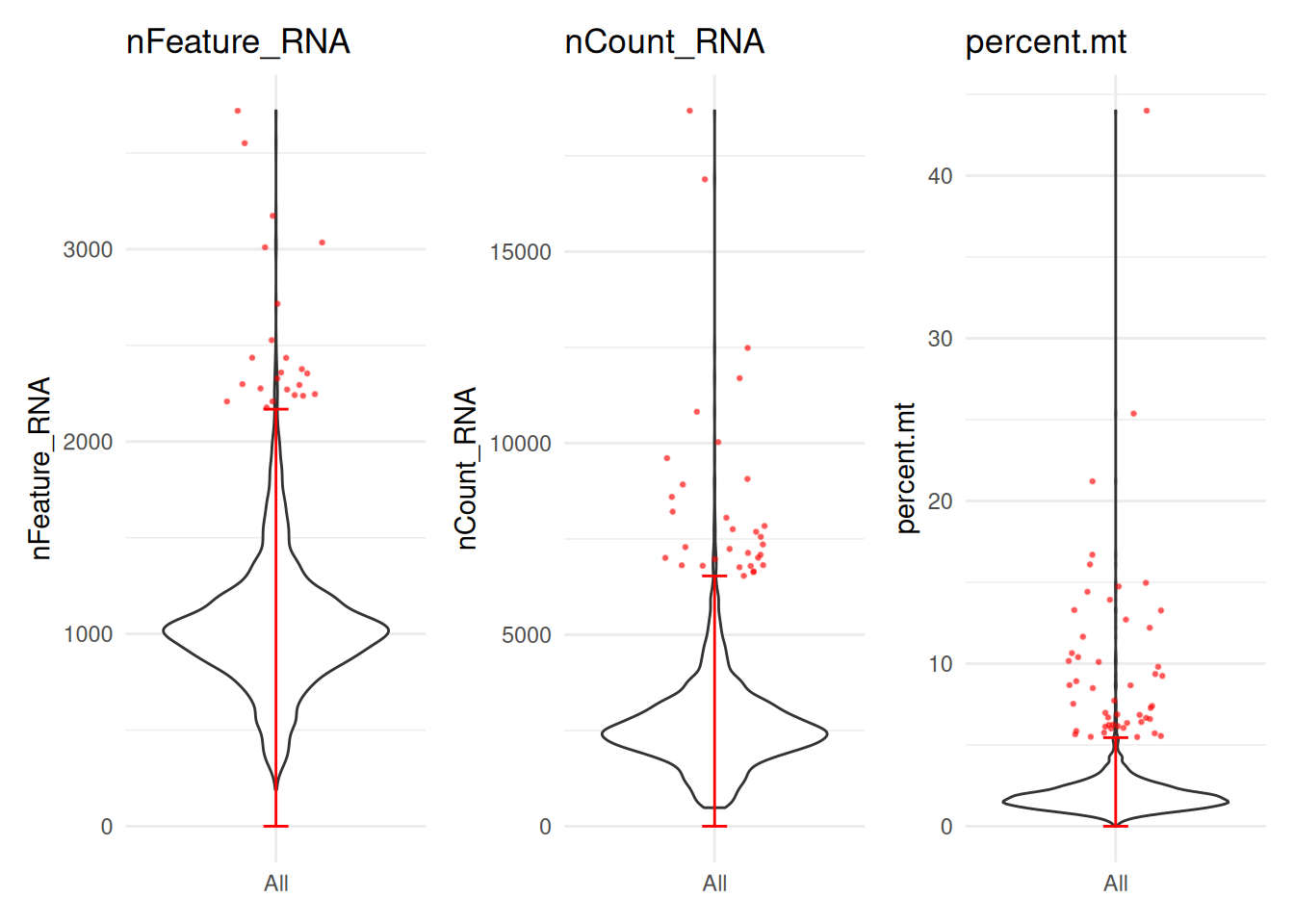
seurat_obj <- seurat_obj |> sn_filter_genes()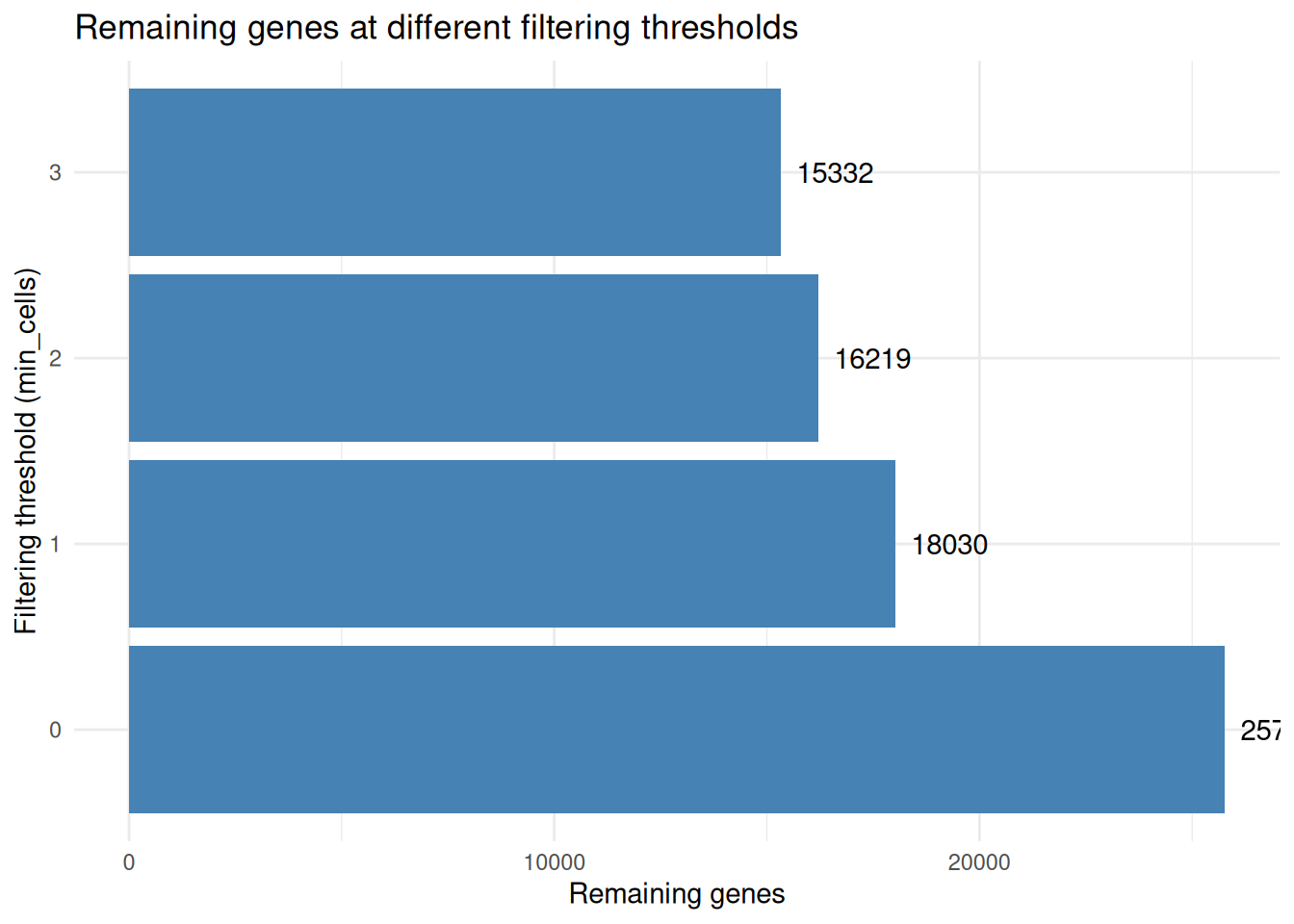
R
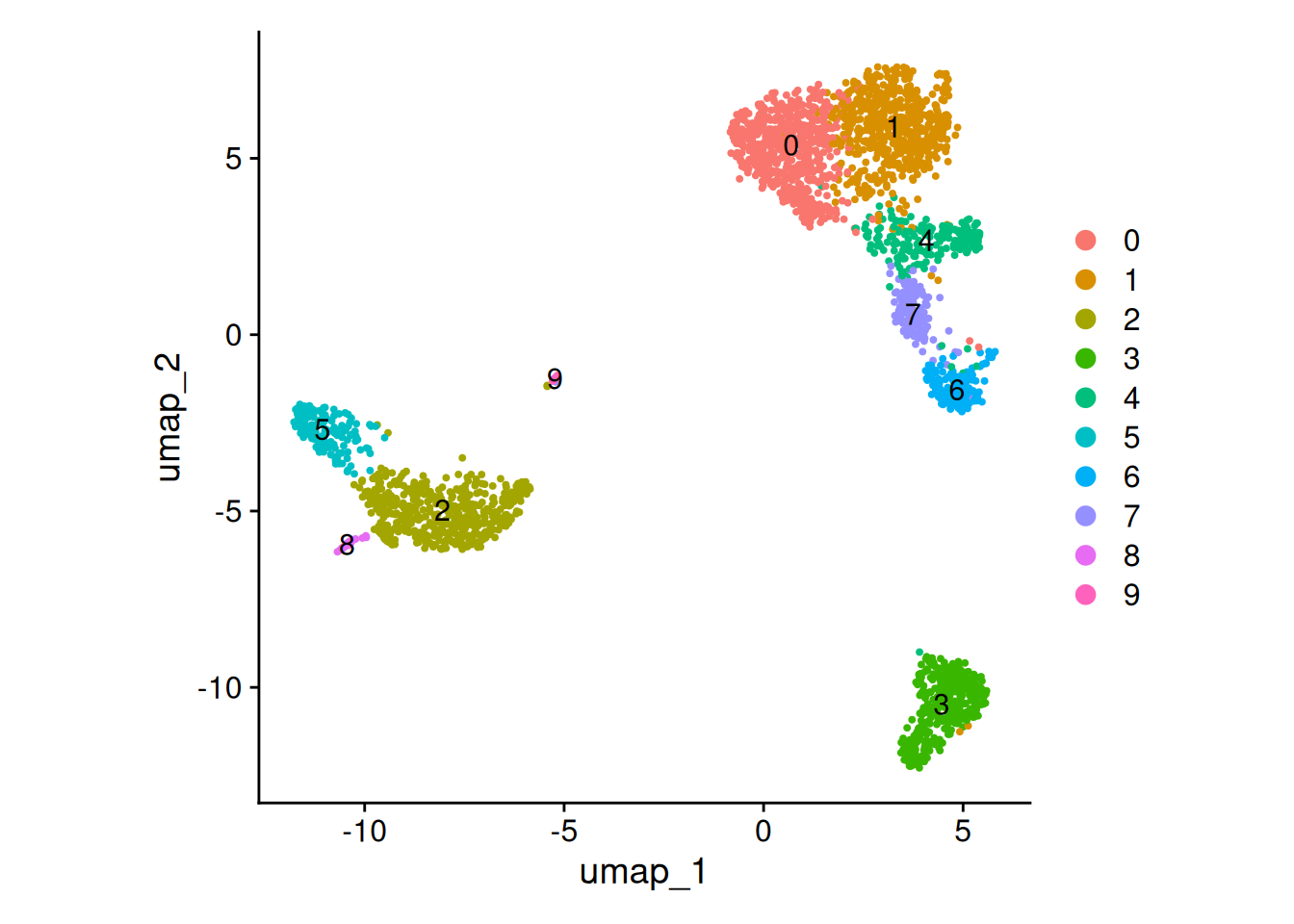
seurat_obj <- seurat_obj |>
sn_run_cluster()DimPlot(object = seurat_obj, reduction = "umap", label = TRUE) +
ggplot2::theme(aspect.ratio = 1)